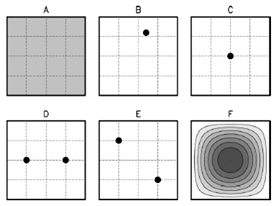
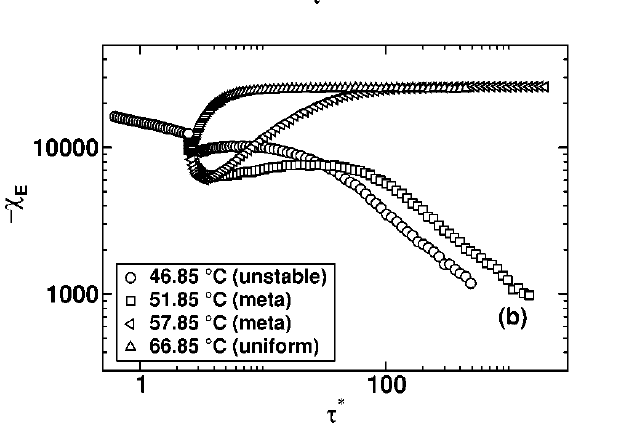
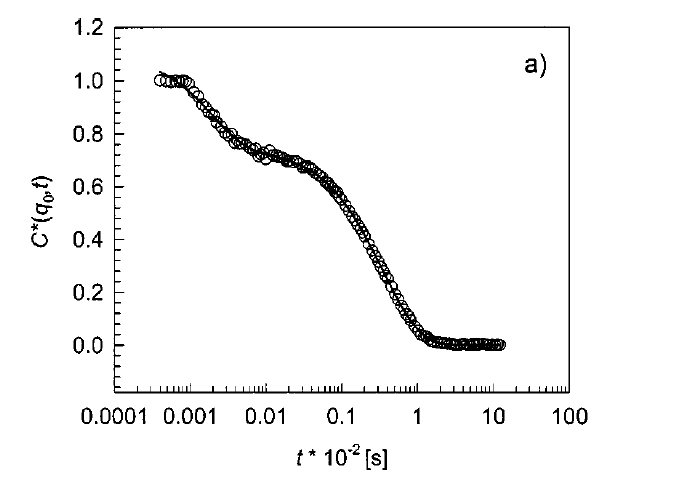
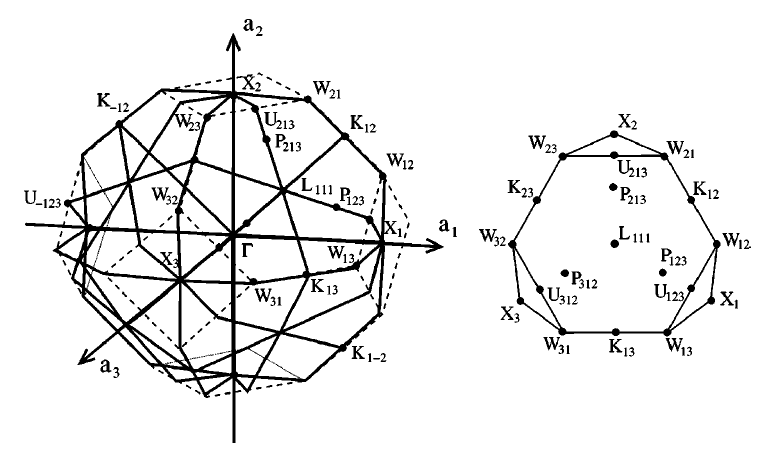
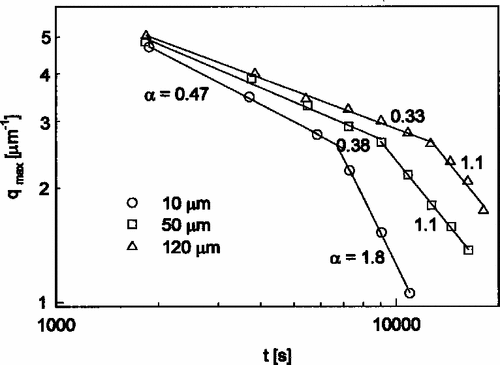
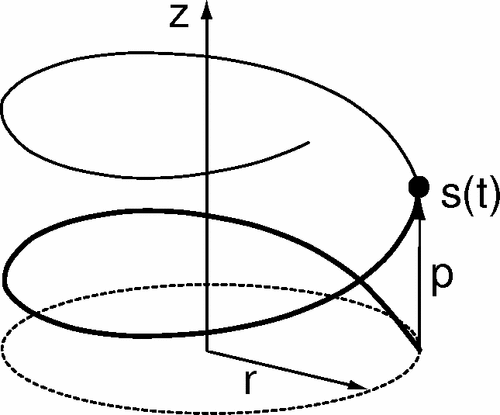
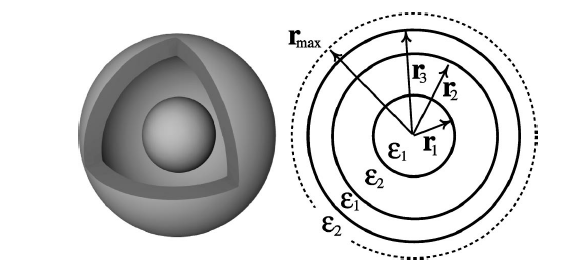
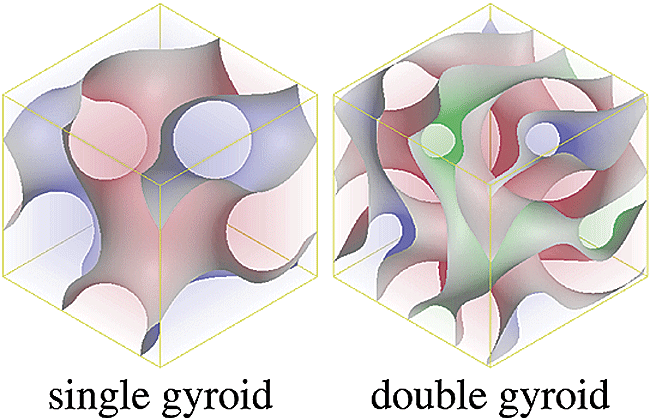
Some features of soft matter systems
R. Hołyst
Soft Matter, 2005,1, 329-333
Pierre Gilles de Gennes was awarded a Nobel prize in physics “for discovering that methods developed for studying order phenomena in simple systems can be generalized to more complex forms of matter, in particular to liquid crystals and polymers”. Thanks to his works “soft matter” became a new legitimate discipline in physics. Soft matter includes a vast range of materials, which cannot be classified as simple liquids or solids. Many soft matter systems exhibit partially broken translational and/or rotational symmetry. In others we observe mesoscopic self-assembling into supramolecular structures leading to viscoelastic behavior. The partial ordering with viscoelastic properties, topological and geometrical complexity, and long relaxations associated with broken symmetries and/or supramolecular assembling are the main features of these systems. Among them we find liquid crystals, gels, biological membranes, colloidal suspensions, polymer solutions and polymer melts and blends, surfactant solutions etc. Typical models used in soft matter theory are based on statistical mechanics and classical thermodynamics, supplemented by the theory of elasticity, hydrodynamics and thermodynamics of irreversible processes and also some elements of the field theory. In this short overview I would like to discuss three theoretical issues related to soft matter systems: interactions, the role of the entropy, and finally the order parameter description.